Based on the Maui Derm 2018 presentation by Joel M. Gelfand MD, MSCE, FAAD, from University of Pennsylvania Perelman School of Medicine, Philadelphia Pennsylvania
Article by Jo Ann LeQuang and the Journal of Clinical and Aesthetic Dermatology
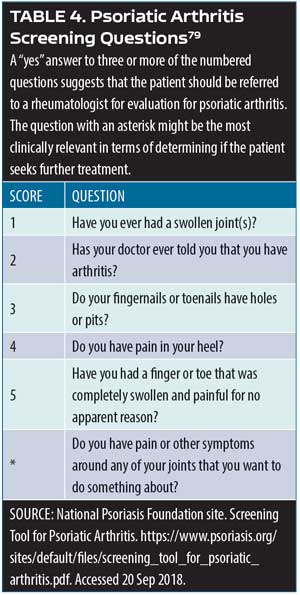
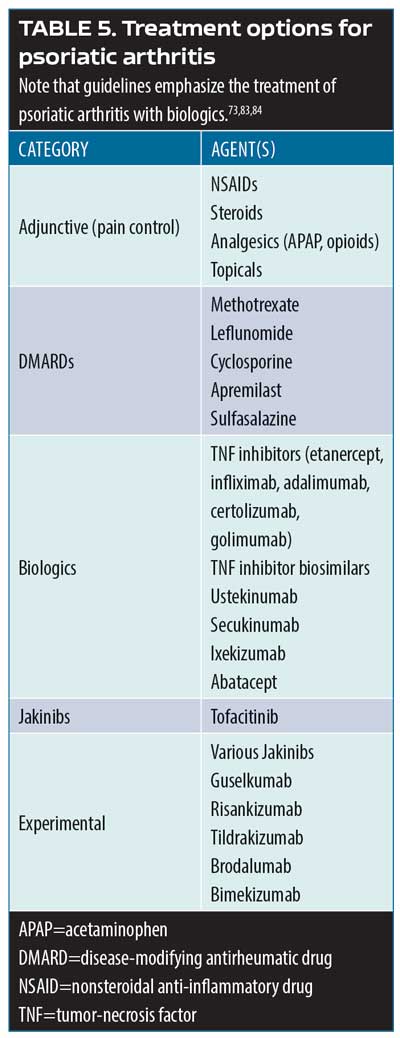
Medical literature on the topic of psoriasis comorbidities has been rapidly expanding since around the turn of the century. There is a strong body of evidence that patients with psoriasis are at increased risk for developing psoriatic arthritis, cardiovascular disease (e.g., myocardial infarction, stroke), Crohn’s disease, diabetes, metabolic syndrome, mood disorders (e.g., anxiety, depression, suicide), and a much rarer condition, T-cell lymphoma.7–14 Genes and loci associated with psoriasis overlap with those associated with other conditions, such as diabetes and cardiovascular diseases (PSORS2/3/4, CDKAL1, ApoE4, TNFAIP32), with the potential contribution of environmental risk factors such as obesity and smoking.
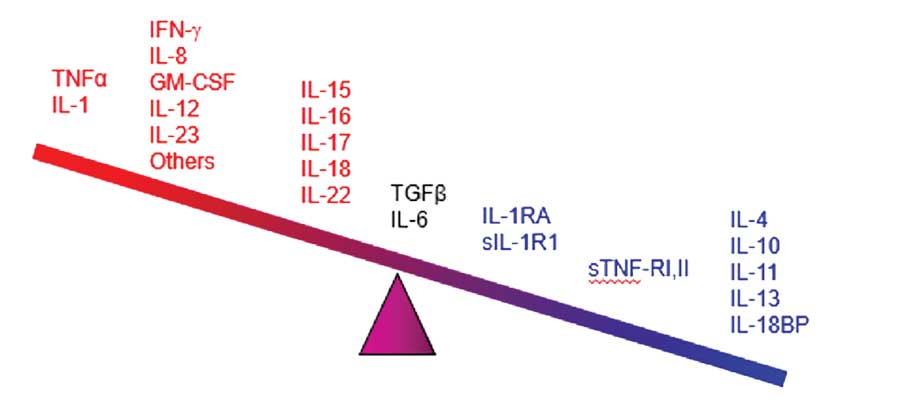
The pathophysiology of psoriasis includes an increased presence of antigens, activation of T-cells and T-helper cell Types 1 and 17 cytokines (Th1 and Th17), and possibly higher levels of C-reactive proteins. These immunological conditions make patients vulnerable to other diseases associated with inflammatory processes.7–13 Chronic inflammation has been associated with numerous conditions, such as atherosclerosis and thrombosis.9 Epidermal proliferation associated with psoriasis plaques can increase uric acid and exacerbate oxidative stress.7 Patients under treatment for psoriasis might be exposed to varying risks and benefits associated with drug therapies.11 Finally, psoriasis in and of itself is associated with an adverse psychosocial impact owing to its disfiguring nature and chronic persistence.14 All of these factors combine such that psoriasis is comorbid with numerous conditions.
Cardiometabolic diseases. In a four-year, population-based cohort study from the United Kingdom (n=8,124 patients with psoriasis and 76,599 controls),15 patients with psoriasis had a hazard ratio for development of Type II diabetes mellitus of 1.64 (95% CI, 1.23– 2.18) if more than 10 percent of their BSA was affected. For each 10-percent increase in BSA affected by psoriasis, there was a 20-percent increase in risk for developing diabetes. This is not trivial: about 125,650 new cases of diabetes every year can be directly linked to psoriasis.15
Cardiovascular diseases. A prospective database study (n=130,976 patients with psoriasis and 556,995 control patients) of 5.4 years duration found patients with psoriasis had a higher rate of myocardial infarction than controls and exhibited a dose-response effect, in that those with the most severe psoriasis had the highest rate of myocardial infarction.7 A cohort study using the General Practice Research Database (N=3,603 patients with psoriasis) found that patients with severe psoriasis were at increased risk for cardiovascular death, independent of the traditional cardiovascular risk factors.16 Recent studies suggest that elevated risk for cardiovascular conditions might be attributed to arterial stiffness, an early independent predictor of adverse cardiovascular events. Arterial stiffness occurs frequently among psoriasis patients.17
Stroke. Stroke is a major cause of morbidity and mortality, and its associations with diabetes, hypertension, atrial fibrillation, transient ischemic attacks, and smoking are well known.18 In a population-based cohort study, it was found that severe psoriasis increased an individual’s chance of having a stroke by 44 percent. There is an elevated risk for stroke for all patients with psoriasis, but the risk for patients with mild psoriasis was deemed modest.9
Cancer. Patients with psoriasis are at an increased risk of cancer, particularly, but not exclusively, NMSC. It remains unclear whether new psoriasis treatments targeting interleukin (IL) 12 and 23 affect the patient’s risk for cancer.19 IL-12 and IL-23 play a role in the cellular responses and inflammatory processes associated with psoriasis but they are also involved in the immune response to tumors and infections. A systematic review and meta-analysis found that individuals with a history of cancer who developed psoriasis (or other immune-related disorders such as rheumatoid arthritis and inflammatory bowel disease) and were treated with TNF inhibition did not show an elevated risk for cancer recurrence or a new cancer.20 A review of cancer rates among patients with various immune-mediated diseases found that there was not an increased rate of cancer among patients with psoriasis in general or among patients with psoriasis who were on TNF inhibition therapy.21
Liver disease. Patients with psoriasis are at increased risk for all liver disease, nonalcoholic fatty liver disease (NAFLD), and cirrhosis. The prevalence of liver disease and cirrhosis was shown to increase in proportion to the BSA affected by psoriasis.22 The prevalence of NAFLD is estimated to be 20 to 60 percent among patients with plaque psoriasis.23–27 Nonalcoholic steatohepatitis (NASH) is a progressive form of fatty liver disease with an incidence of about 22 percent among patients with psoriasis.26 This area warrants further study.
Emerging comorbidities. Infections. A new area of research explores the risk of serious infection and opportunistic infection in patients with psoriasis.28 The risks of opportunistic infections and herpes zoster have been shown to be elevated among patients with moderate-to-severe psoriasis and could be statistically associated with immunosuppressive therapy.29
Sleep disorders. Psoriasis adversely affects sleep and has been associated with comorbid obstructive sleep apnea.30 It is thought that intermittent hypoxia caused by sleep apnea along with sleep deprivation creates a chronic inflammatory state that can set the stage for autoimmune disorders such as psoriasis.31
Chronic obstructive pulmonary disease (COPD). COPD shares some risk factors with psoriasis, including obesity, smoking, a sedentary lifestyle, and an association with metabolic syndrome, as well as potentially shared biological pathways such as Th17 driven inflammation.32 Patients with psoriasis have increased risk for impaired pulmonary function compared with control patients, as evidenced on spirometry.33 Patients with psoriasis also have a greater risk of developing COPD, compared with the general population, with an OR of 1.90 (95% CI, 1.36–2.65), and this risk increases with the severity of the psoriasis (2.15 for severe psoriasis, 95% CI, 1.26–3.67).34
Chronic kidney disease and end-stage renal disease (ESRD). Chronic kidney disease and ESRD have also been associated with psoriasis, according to a systematic review and meta-analysis (n=199,808), with an estimated pooled risk ratio for patients with psoriasis and chronic kidney disease of 1.34 (95% CI, 1.14–1.57) and psoriasis and ESRD of 1.29 (95% CI, 1.05–1.60).35 Severe psoriasis confers a greater risk than milder forms of psoriasis, with an adjusted hazard ratio of 1.90 (95% CI, 1.33–2.70) for chronic kidney disease and 2.97 (95% CI, 1.72–5.11) for ESRD.36
Peptic ulcer disease. Patients with psoriasis are at a greater risk than patients without psoriasis for developing peptic ulcer disease (adjusted OR 1.27, 95% CI, 1.03–1.58).37
Sexual dysfunction. Patients with psoriasis are more likely than the general population to experience sexual dysfunction, in particular erectile dysfunction (ED).38 According to one study,39 prevalence of ED was much higher in male patients with psoriasis (n=135, 61.5%) compared with controls (n=201, 48.3%), (p=0.001).
Drug therapies. TNF inhibitors. The systemic inflammation associated with psoriasis, psoriatic arthritis, and rheumatoid arthritis accelerates atherosclerosis and, in this way, might promote cardiovascular disorders. This risk exists even in patients with psoriasis who have no other cardiovascular risks, and cardiovascular disorders are especially prevalent in those with psoriasis and other cardiovascular risk factors. Thus, treatment of the underlying inflammation has been proposed to mitigate the cardiovascular risk.40 In a systematic review on rheumatoid arthritis,41 TNF inhibitors, such as infliximab, etanercept, and adalimumab, have been shown to be cardioprotective. In a meta-analysis of studies assessing the effect of TNF inhibitors on adverse cardiovascular events in patients with psoriasis, with or without psoriatic arthritis (n=49,795, 5 studies),42 the risk for a cardiovascular adverse event was significantly lower when patients were treated with TNF inhibitors compared with topical treatments or phototherapy (risk ratio 0.58, 95% CI, 0.43–0.77, p<0.001). In this meta-analysis,42 278 patients in the TNF inhibitor group and 1,063 in the control group had suffered myocardial infarctions. TNF inhibitors appeared to significantly reduce cardiovascular adverse events compared with methotrexate (risk ratio 0.67, 95% CI, 0.52–0.88, p=0.003). The risk for myocardial infarction specifically was significantly decreased, compared with topical therapy or phototherapy, among patients treated with TNF inhibition (risk ratio 0.73, 95% CI, 0.59–0.90, p=0.003) or with methotrexate (risk ratio 0.65, 95% CI, 0.48–0.89, p=0.007). The role of TNF inhibitors compared with other treatments for all-cause mortality and stroke remains to be elucidated. It must be noted that TNF inhibitors do not reduce the rate of heart failure and, in fact, might worsen it.43–45
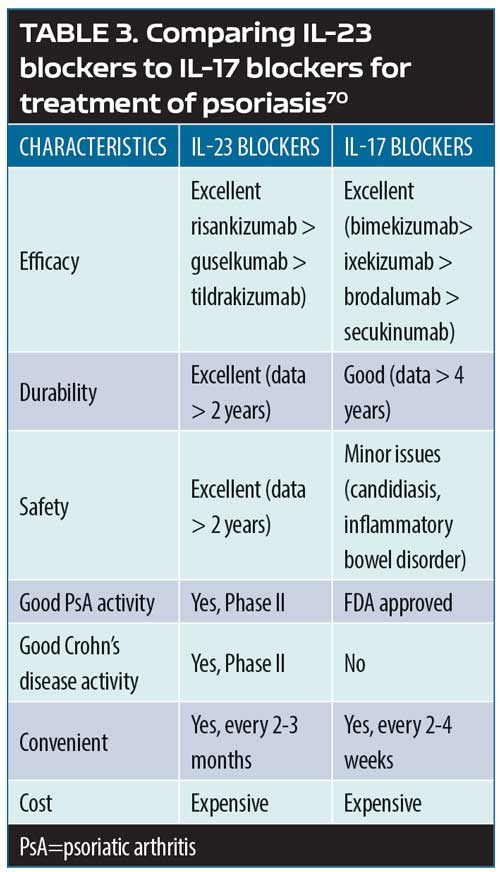
Biologic therapies. In a systematic review and meta-analysis of 38 randomized clinical trials of adults with plaque psoriasis (n=18,024),46 the use of biologic therapies overall was not associated with any significant differences in the risk for major adverse cardiovascular events (MACEs), with an OR of 1.45 and 95-percent CI of 0.34–6.24. Specifically, for TNF alpha inhibitors (adalimumab, etanercept, infliximab), the OR was 0.67 (95% CI, 0.10–4.63); for anti-IL-17A agents (secukinumab and ixekizumab), the OR was 1.00 (95% CI, 0.09–11.09), and ustekinumab had an OR of 4.48 (95% CI,
0.24–84.77). There were no statistically significant differences among these various agents at approved doses compared with placebo.46
In light of what we know now, how should psoriasis be treated? People with psoriasis face a greater mortality burden compared with the general population.47 In a study from Noe and colleagues,48 BSA affected by psoriasis conferred a mortality risk such that patients with more than 10-percent affected BSA had a higher risk of all-cause death than those with less than three-percent BSA involvement.48 For many conditions, the more severe the psoriasis, the greater the comorbid risk to the patient. Thus, moderate and severe cases of psoriasis warrant special consideration.
Since patients with psoriasis are at increased risk for a number of cardiovascular conditions, dermatologists should educate these patients with psoriasis about the increased risk for cardiovascular disease and screen them according to standard guidelines for hypertension, diabetes, and hyperlipidemia. At present, fewer than half of all dermatologists ever conduct such screenings, but it is important since these patients might not be seeing other physicians or these other physicians might be unaware of a patient’s psoriasis and the risk it poses.49–51 Thirty-seven percent of newly screened patients with psoriasis were found to be at high risk for cardiovascular disease, and almost half of them (46%) were not optimally managed for these risk factors.49–51 The question that emerges from our growing knowledge of psoriasis comorbidities is whether more aggressive treatment of psoriasis would lower the risk of cardiovascular disease, cardiometabolic disorders, and overall mortality. Moreover, which type(s) of pharmacological therapy would be most helpful in reducing the risk of MACE and cardiovascular mortality? Early observational data suggest that methotrexate52,53 and TNF inhibitors54–56 are able to lower the risk of certain cardiovascular events (but not heart failure). Data are not yet available on whether phototherapy, topical treatments, and specific drugs such as apremilast, ustekinumab, secukinumab, ixekizumab, and guselkumab confer any cardioprotective benefits to patients with psoriasis. Randomized, controlled clinical studies and further systematic reviews and meta-analyses are needed to guide prescribers with more definitive evidence.
Treatment of atherosclerosis focuses mainly on reducing plasma lipid levels, but biomarkers for inflammation (high-sensitivity C-reactive protein and IL-6) have likewise been associated with an elevated risk for cardiovascular events independent of cholesterol level. Since statin drugs reduce both cholesterol levels and inflammation, it is unclear whether the reduction of vascular inflammation alone (without concomitant reduction of lipids) can reduce cardiovascular events associated with atherosclerosis. Canakinumab is a fully human monoclonal antibody that targets IL-1 beta and has known anti-inflammatory effects. The Canakinumab Anti-inflammatory Thrombosis Outcome Study (CANTOS)57 was a randomized, double-blind, placebo-controlled clinical trial enrolling 10,061 patients with a prior myocardial infarction in order to determine if canakinumab could prevent recurrent vascular events in patients with a high-sensitivity C-reactive protein level of 2mg/L or greater. At 48 months, the median reduction from baseline in C-reactive protein levels was 26-percent, 37-percent, and 41-percent greater in patients who received 50, 150, or 300mg of canakinumab, respectively, compared with placebo.57 IL-6 levels also decreased, measured at 12 months. Canakinumab had no significant effect on either low-density lipoproteins (LDL) or high-density lipoproteins (HDL) cholesterol levels and resulted in a median of about five-percent elevation in triglycerides. The primary endpoint of the study was a composite endpoint of nonfatal myocardial infarction, nonfatal stroke, or cardiovascular death; the composite endpoint occurred significantly less often among the 150mg canakinumab group compared with placebo (p=0.0275). Patients in the canakinumab group were more likely to experience neutropenia than those in the placebo group, and the pooled canakinumab group had significantly higher mortality attributable to infection or sepsis (incidence rate was 0.31 compared to 0.18 per 100 person-years, p=0.02).55 IL-1 beta inhibition appears to be an important anti-inflammatory pathway that might offer a degree of cardioprotection, but it is associated with its own risks for infection.57
Other studies are ongoing in evaluating the role of vascular inflammation and lipid metabolism in moderate-to-severe psoriasis. The Cardiovascular Inflammation Reduction Trial (CIRT)58 will evaluate methotrexate’s role in the possible reduction of major vascular events in a population without psoriasis but with a history of myocardial infarction, diabetes, or metabolic syndrome. TNF inhibition does not appear to reduce vascular inflammation compared with placebo at 16 weeks.59 The PSOLAR data do not show that biologics reduce the risk of MACE in patients with moderate-to-severe psoriasis.60
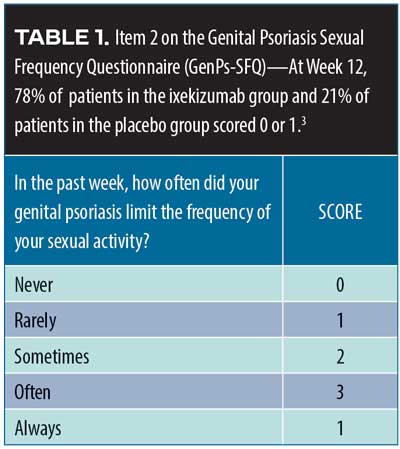
The association between psoriasis and cancer exists but remains to be more fully elucidated. As a precautionary measure, patients with psoriasis should be advised to stay current with all age-appropriate cancer screenings (e.g., colon, cervical breast, prostate, and lung). The incidence of smoking is higher among patients with psoriasis than the general population, so low-dose computed tomography (CT) screenings for lung cancer might be appropriate for patients 55 to 80 years of age with a history of smoking 30 or more packs of cigarettes per year as well as for current smokers and smokers who have quit in the last 15 years. Since psoriasis is associated with sexual dysfunction, clinicians might wish to ask patients about their sexual health.61 Lesions in the genital area can be particularly distressing and should be discussed with patients.62 Since over 45 percent of patients with genital-area psoriasis never discuss the problem with a clinician, the dermatologist might need to initiate such conversations.63 Patients with psoriasis are at increased risk for infections, so patients with guttate flares should be screened for possible streptococcal infection. Patients with severe psoriasis should also be screened for human immunodeficiency virus (HIV). Patients with psoriasis should be regularly vaccinated for influenza, pneumonia, hepatitis B, human papilloma virus (ages 9– 26 years), and shingles (>50 years of age).64
References
7. Gelfand JM, Neimann AL, Shin DB, et al. Risk of myocardial infarction in patients with psoriasis. J Am Med Assoc. 2006;296(14):1735–1741.
8. Gelfand JM, Azfar RS, Mehta NN. Psoriasis and cardiovascular risk: strength in numbers. J Invest Dermatol. 2010;130(4):919–922.
9. Gelfand JM, Dommasch ED, Shin DB, et al. The risk of stroke in patients with psoriasis. J Invest Dermatol. 2009;129(10):2411–2418.
10. Gelfand JM, Yeung H. Metabolic syndrome in patients with psoriatic disease. J Rheumatol. 2012;89:24–28.
11. Lee MS, Lin RY, Lai MS. Increased risk of diabetes mellitus in relation to the severity of psoriasis, concomitant medication, and comorbidity: a nationwide population-based cohort study. Journal of the American Academy of Dermatology. 2014;70(4):691-698.
12. Takeshita J, Grewal S, Langan SM, et al. Psoriasis and comorbid diseases: Implications for management. Journal of the American Academy of Dermatology. 2017;76(3):393-403.
13. Takeshita J, Grewal S, Langan SM, et al. Psoriasis and comorbid diseases: epidemiology. J Am Acad Dermatol. 2017;76(3):377–390.
14. Connor CJ, Liu V, Fiedorowicz JG. Exploring the physiological link between psoriasis and mood disorders. Dermatol Res Prac. 2015;2015:409637.
15. Wan MT, Shin DB, Hubbard RA, et al. Psoriasis and the risk of diabetes: a prospective population-based cohort study. J Am Acad Dermatol. 2018;78(2):315–322.e311.
16. Mehta NN, Azfar RS, Shin DB, et al. Patients with severe psoriasis are at increased risk of cardiovascular mortality: cohort study using the General Practice Research Database. Eur Heart J. 2010;31(8):1000–1006.
17. Dregan A. Arterial stiffness association with chronic inflammatory disorders in the UK Biobank study. Heart (British Cardiac Society). 2018;104(15):1257–1262.
18. Donnan GA, Fisher M, Macleod M, Davis SM. Stroke. Lancet. 2008;371(9624):1612–1623.
19. Ergen EN, Yusuf N. Inhibition of interleukin 12 and/or interleukin 23 for treatment of psoriasis: What is the evidence for an effect on malignancy? Exp Dermatol. 2018;27(7):737–747.
20. Micic D, Komaki Y, Alavanja A, et al. Risk of cancer recurrence among individuals exposed to antitumor necrosis factor therapy: a systematic review and meta-analysis of observational studies. J Clin Gastroenterol. 2017 Jul 21. doi: 10.1097/MCG.0000000000000865. [Epub ahead of print].
21. Chen Y, Friedman M, Liu G, et al. Do tumor necrosis factor inhibitors increase cancer risk in patients with chronic immune-mediated inflammatory disorders? Cytokine. 2018;101:
78–88.
22. Ogdie A, Grewal SK, Noe MH, et al. Risk of incident liver disease in patients with psoriasis, psoriatic arthritis, and rheumatoid arthritis: a population-based study. J Invest Dermatol. 2018;138(4):760–767.
23. Gisondi P, Targher G, Zoppini G, Girolomoni G. Non-alcoholic fatty liver disease in patients with chronic plaque psoriasis. J Hepatol. 2009;51(4):758–764.
24. Miele L, Vallone S, Cefalo C, et al. Prevalence, characteristics and severity of non-alcoholic fatty liver disease in patients with chronic plaque psoriasis. J Hepatol. 2009;51(4):778–786.
25. Madanagobalane S, Anandan S. The increased prevalence of non-alcoholic fatty liver disease in psoriatic patients: a study from South India. Australas J Dermatol. 2012 Aug;53(3):190–197.
26. Roberts KK, Cochet AE, Lamb PB, et al. The prevalence of NAFLD and NASH among patients with psoriasis in a tertiary care dermatology and rheumatology clinic. Aliment Pharmacol Ther. 2015 Feb;41(3):293–300.
27. van der Voort EA, Koehler EM, Dowlatshahi EA, et al. Psoriasis is independently associated with nonalcoholic fatty liver disease in patients 55 years old or older: results from a population-based study. J Am Acad Dermatol. 2014;70(3):517–524.
28. Wakkee M, de Vries E, van den Haak P, Nijsten T. Increased risk of infectious disease requiring hospitalization among patients with psoriasis: a population-based cohort. J Am Acad Dermatol. 2011;65(6):1135–1144.
29. Takeshita J, Shin DB, Ogdie A, Gelfand JM. Risk of serious infection, opportunistic infection, and herpes zoster among patients with psoriasis in the United Kingdom. J Invest Dermatol. 2018;138(8):1726–1735.
30. Callis Duffin K, Wong B, Horn EJ, Krueger GG. Psoriatic arthritis is a strong predictor of sleep interference in patients with psoriasis. J Am Acad Dermatol. 2009;60(4):604–608.
31. Vakil M, Park S, Broder A. The complex associations between obstructive sleep apnea and auto-immune disorders: a review. Med Hypotheses. 2018;110:138–143.
32. Machado-Pinto J, Diniz Mdos S, Bavoso NC. Psoriasis: new comorbidities. An Bras Dermatol. 2016;91(1):8–14.
33. Balci DD, Celik E, Genc S, Celik MM, Inan MU. Impaired pulmonary function in patients with psoriasis. Dermatology (Basel, Switzerland). 2016;232(6):664–667.
34. Li X, Kong L, Li F, et al. Association between psoriasis and chronic obstructive pulmonary disease: a systematic review and meta-analysis. PloS One. 2015;10(12):e0145221.
35. Ungprasert P, Raksasuk S. Psoriasis and risk of incident chronic kidney disease and end-stage renal disease: a systematic review and meta-analysis. Int Urol Nephrol. 2018;50(7):1277–1283.
36. Chi C, Wang J, Chen Y, Wang S, et al. Risk of incident chronic kidney disease and end-stage renal disease in patients with psoriasis: a nationwide population-based cohort study. J Dermatol Sci. 2015 Jun;78(3):232–238
37. Yeung H, Takeshita J, Mehta NN, et al. Psoriasis severity and the prevalence of major medical comorbidity: a population-based study. JAMA Dermatol. 2013;149(10):1173–1179.
38. Molina-Leyva A, Jimenez-Moleon JJ, Naranjo-Sintes R, Ruiz-Carrascosa JC. Sexual dysfunction in psoriasis: a systematic review. J Eur Acad Dermatol Venereol. 2015;29(4):649–655.
39. Cabete J, Torres T, Vilarinho T, et al. Erectile dysfunction in psoriasis patients. Eur J Dermatol. 2014 ;24(4):482–486
40. Smolen JS, Landewe R, Breedveld FC, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2013 update. Ann Rheum Dis. 2014;73(3):492–509
41. Roubille C, Richer V, Starnino T, et al. The effects of tumour necrosis factor inhibitors, methotrexate, non-steroidal anti-inflammatory drugs and corticosteroids on cardiovascular events in rheumatoid arthritis, psoriasis and psoriatic arthritis: a systematic review and meta-analysis. Ann Rheum Dis. 2015;74(3):480–489.
42. Yang ZS, Lin NN, Li L, Li Y. The effect of TNF inhibitors on cardiovascular events in psoriasis and psoriatic arthritis: an updated meta-analysis. Clin Rev Allergy Immunol. 2016 Oct;51(2):240-7.
43. Levine B, Kalman J, Mayer L, et al. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. New Eng J Med. 1990;323(4):236–241.
44. Kadokami T, Frye C, Lemster B, et al. Anti-tumor necrosis factor-alpha antibody limits heart failure in a transgenic model. Circulation. 2001;104(10):1094–1097.
45. Chung ES, Packer M, Lo KH, et al. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation. 2003;107(25):
3133–3140.
46. Rungapiromnan W, Yiu ZZN, Warren RB, et al. Impact of biologic therapies on risk of major adverse cardiovascular events in patients with psoriasis: systematic review and meta-analysis of randomized controlled trials. Br J Dermatol. 2017;176(4):890–901.
47. Ogdie A, Haynes K, Troxel AB, et al. Risk of mortality in patients with psoriatic arthritis, rheumatoid arthritis and psoriasis: a longitudinal cohort study. Ann Rheum Dis. 2014 Jan;73(1):
149-153.
48. Noe MH, Shin DB, Wan MT, Gelfand JM. Objective measures of psoriasis severity predict mortality: a prospective population-based cohort study. J Invest Dermatol. 2018;138(1):
228–230.
49. Alamdari HS, Gustafson CJ, Davis SA, et al. Psoriasis and cardiovascular screening rates in the United States. J Drugs Dermatol. 2013;12(1):
e14–19.
50. Manalo IF, Gilbert KE, Wu JJ. Survey of trends and gaps in dermatologists’ cardiovascular screening practices in psoriasis patients: areas still in need of improvement. J Am Acad Dermatol. 2015;73(5):872-874.
51. Rutter MK, Kane K, Lunt M, et al. Primary care-based screening for cardiovascular risk factors in patients with psoriasis. Br J Dermatol. 2016;175(2):348–356.
52. Micha R, Imamura F, Wyler von Ballmoos M, et al. Systematic review and meta-analysis of methotrexate use and risk of cardiovascular disease. Am J Cardiol. 2011;108(9):1362–1370.
53. Prodanovich S, Ma F, Taylor JR, Pezon C, Fasihi T, Kirsner RS. Methotrexate reduces incidence of vascular diseases in veterans with psoriasis or rheumatoid arthritis. J Am Acad Dermatol. 2005;52(2):262–267.
54. Barnabe C, Martin BJ, Ghali WA. Systematic review and meta-analysis: anti-tumor necrosis factor alpha therapy and cardiovascular events in rheumatoid arthritis. Arthritis Care Res (Hoboken). 2011;63(4):522–529.
55. Wu JJ, Poon KY, Channual JC, Shen AY. Association between tumor necrosis factor inhibitor therapy and myocardial infarction risk in patients with psoriasis. Arch Dermatol. 2012;148(11):1244–1250.
56. Ahlehoff O, Skov L, Gislason G, et al. Cardiovascular disease event rates in patients with severe psoriasis treated with systemic anti-inflammatory drugs: a Danish real-world cohort study. J Intern Med. 2013;273(2):197–204.
57. Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory Tterapy with canakinumab for atherosclerotic disease. New Engl J Med. 2017;377(12):1119–1131.
58. Everett BM, Pradhan AD, Solomon DH, et al. Rationale and design of the Cardiovascular Inflammation Reduction trial: a test of the inflammatory hypothesis of atherothrombosis. Am Heart J. 2013;166(2):199–207.e115.
59. Bissonnette R, Harel F, Krueger JG, et al. TNF-alpha antagonist and vascular inflammation in patients with psoriasis vulgaris: a randomized placebo-controlled study. J Invest Dermatol. 2017;137(8):1638–1645.
60. Bissonnette R, Kerdel F, Naldi L, et al. Evaluation of risk of major adverse cardiovascular events with biologic therapy in patients with psoriasis. J Drugs Dermatol. 2017;16(10):1002–1013.
61. Chen YJ, Chen CC, Lin MW, et al. Increased risk of sexual dysfunction in male patients with psoriasis: a nationwide population-based follow-up study. J Sex Med. 2013;10(5):1212–1218.
62. Meeuwis KA, de Hullu JA, van de Nieuwenhof HP, et al. Quality of life and sexual health in patients with genital psoriasis. Br J Dermatol. 2011;164(6):1247–1255.
63. Meeuwis KA, van de Kerkhof PC, Massuger LF, et al. Patients’ experience of psoriasis in the genital area. Dermatology. 2012;224(3):271–276.
64. United States Centers for Disease Control and Prevention site. Recommended Immunization Schedule for Adults Aged 19 Years or Older, United States 2018. 6 Feb 2018. https://www.cdc.gov/vaccines/schedules/hcp/adult.html. Accessed 24 Sep 2018.