Update on the Pathogenesis of Psoriasis: A Bench-to-Bedside Success Story
Based on the Maui Derm 2018 presentation by Andrew Blauvelt, MD, MBA, Oregon Medical Research Center, Portland, Oregon
Article by Jo Ann LeQuang and the Journal of Clinical and Aesthetic Dermatology
Psoriasis is caused by the complex interplay of four factors: autoantigens, environmental antigens, genetics, and the immune system. Autoinflammation, an inflammatory state not mediated by circulating autoantibodies and autoreactive T-cells, has recently been implicated as well.65 Genetics remains the key to better understanding the immune system and the pathogeneses of all autoimmune disorders, including psoriasis. Better understanding of the pathogenesis of psoriasis will allow for more accurate mapping of psoriasis pathways and improved ability to identify therapeutic targets, which, in turn, will lead to safer and more effective treatments.
The genetics of psoriasis. Psoriasis has strong heritability, and over 70 susceptibility loci/genes associated with psoriasis have been identified to date.66 In monozygotic twins, there is approximately a 70-percent genetic concordance, and 10 percent of the children of parents with psoriasis will develop psoriasis themselves (a 4- to 5-fold increase over general population). In the 1970s, psoriasis was found to be associated with the HLA Class I antigens, with associations between psoriasis and HLA-C (HLA-Cw6 in particular) and HLA-B alleles.67 The most common and dominant gene association is HLA-C*0602, but only 41 percent of people with psoriasis are HLA-C*0602-positive.68 In fact, the HLA-C*0602 gene, in and of itself, is neither necessary nor sufficient to develop psoriasis. About 20 percent of the population are carriers of the HLA-C*0602 gene, and about 10 percent of carriers develop psoriasis.66
Autoantigens. Antigens are molecules that induce an immune response in a host organism, and antibodies are the body’s method to target these antigens. Antigens are typically peptides, polysaccharides, and lipids that originate endogenously or exogenously. Autoantigens are endogenous antigens that are produced by normal cell metabolism or as a result of a viral or bacterial infection. The major histocompatibility (MHC) Class I antigen pathway serves to alert the immune system of the presence of a cell with a viral infection. MHC molecules are expressed on nucleated cells and platelets. In humans, HLA-A, HLA-B, and HLA-C correspond to MHC Class I, while HLA-DP, HLA-DM, HLA-DOA, HLA-DOB, HLA-DQ, and HLA-DR correspond to MHC Class II. Thus, autoantigens are endogenous proteins that can be attacked by the immune system, which is the reason they are associated with autoimmune disorders.
HLA-C*0602 is an MHC Class I molecule that normally presents autoantigens to CD8 T-cells. The autoantigens are ADAMTSL5 and LL-37. ADAMTSL5 is a peptide derived from the protein ADAMTSL5 and produced by melanocytes that can bind to HLA-C*0602 and stimulate psoriatic CD8+ T-cells. LL-37 is an antimicrobial peptide produced by keratinocytes, and it contains a VRSRRCLRL-like peptide that stimulates psoriatic T-cells.66
Environmental antigens also play a role in psoriasis. Streptococcal M proteins contain peptides that overlap with hyperproliferative keratin (K16/K17) peptides; these Streptococcal M proteins can stimulate psoriatic T-cells and have been implicated in certain cases of guttate psoriasis as environmental antigens. Yeast proteins are known to contain a VRSRRCLRL-like peptide that can stimulate psoriatic T-cells, and these exogenous antigens have been implicated in cases of beer-exacerbated psoriasis.66
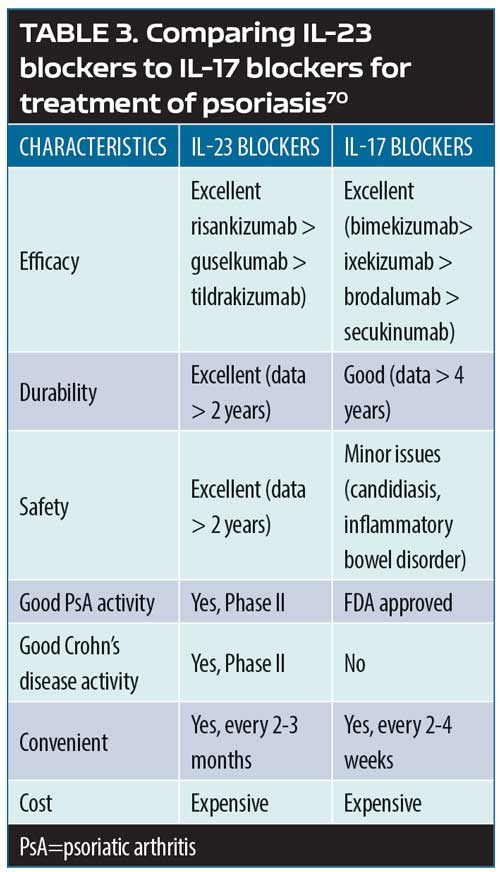
The psoriasis pathways: targets and mechanisms of actions. Psoriasis can be defined as a T-cell-mediated disease mainly driven by pathogenic T-cells that produce high levels IL-17 in response to IL-23. Greater understanding of the IL-23/T-17 axis, identified as the main pathway for psoriasis, has revolutionized the way psoriasis is treated today by offering important targets for pharmacotherapeutic interventions.69 Broadly blocking this pathway (e.g., with methotrexate) might work, but it might not be as safe or effective as a narrower blockade (e.g., TNF inhibition), which targets inflammation more specifically and results in greater efficacy and greater patient safety. However, if the target could be even more narrowly considered—that is, targeting psoriatic inflammation specifically along the IL-23, IL-17A, and IL17RA pathways—this should, in theory at least, allow for even greater safety and better effectiveness. Selective IL-23 blockers include guselkumab, tildrakizumab, and risankizumab. Selective IL-17A blockers include secukinumab, ixekizumab, and bimeizumab. Brodalumab blocks IL-17RA. Thus, pharmacological targets can be viewed as a continuum, with IL-23 the most upstream target and IL-22 and IL-17A downstream. While all of these points represent pharmacologically sound therapeutic targets, there are clinical differences among the inhibitors for these targets (Table 3).69,70
Not all of the cells that produce IL-17A are regulated by IL-23. Some cells, like Th17, depend on IL-23 in order to produce IL-17A, but others produce IL-17A independently of IL-23. This means that IL-23 inhibition will affect some IL-17A producers (those that depend on IL-23) but not the IL-23-independent IL-17A producers. Furthermore, it is thought that some of the IL-17A produced by cells other than Th17 in the skin and the gut might protect the body from candidiasis and inflammatory bowel disease. This would explain the association of candidiasis and inflammatory bowel disorder with IL-17 but not IL-23.69
As a general rule, the pharmacodynamic effects of a drug greatly exceed its pharmacokinetic effects, with the result being that a drug might have an impact on the underlying disease long after it has been eliminated from the body. For example, IL-23 inhibition might also cause the death of pathogenic skin-resident memory T17 cells, which depend on IL-23 for survival. Thus, the prolonged drug effect in destroying these T17 cells might lead to more durable psoriasis control.69
In a study evaluating clinically resolved psoriatic lesions,71 it was found that the lesions contained psoriasis-specific IL-17-producing alpha beta T-cell clones. Healed psoriatic skin (treated with phototherapy and etanercept) showed oligoclonal populations of T-cells, which produce IL-17. This observation might explain why new psoriatic lesions tend to recur at the same site as healed lesions.71
References
66. Elder JT. The quest for psoriasis autoantigens: genetics meets immunology in the melanocyte. J Invest Dermatol. 2017 Oct;137(10):2042–2045.
67. Chandran V. Genetics of psoriasis and psoriatic arthritis. Indian J Dermatol 2010;55(2):151–156.
68. Okada Y, Han B, Tsoi L, et al. Fine mapping of major histocompatibility complex associations in psoriasis and its clinical subtypes. Am J Hum Genet. 2014 Aug 7;95(2):162–72.
69. Hawkes JE, Chan TC, Krueger JG. Psoriasis pathogenesis and the development of novel targeted immune therapies. J Allergy Clin Immunol. 2017;140(3):645–653
70. Maxwell JR, Zhang Y, Brown WA, et al. Differential roles for interleukin-23 and interleukin-17 in intestinal immunoregulation. Immunity. 2015;43(4):739–750.
71. Matos TR, O’Malley JT, Lowry EL, et al. Clinically resolved psoriatic lesions contain psoriasis-specific IL-17-producing alphabeta T cell clones. J Clin Invest. 2017;127(11):4031–4041.