Psoriasis: New Data for 2018
Based on the Maui Derm 2018 presentation by Bruce E. Strober, MD, PhD, Professor and Chair, University of Connecticut Health Center, Department of Dermatology, Farmington, Connecticut
Article by Jo Ann LeQuang and the Journal of Clinical and Aesthetic Dermatology
Four significant studies emerge when perusing the new data from 2017, all of which can help guide clinical care of patients with psoriasis. The first involves treating psoriasis in pregnant and lactating women. Treating psoriasis in any special population can pose clinical challenges, but new and encouraging answers are available for certolizumab pegol. Second, the prevalent and distressing condition of genital psoriasis has always been difficult for patients and prescribers. New 2017 data on ixekizumab as a potential course of treatment holds promise. Third, novel treatments for psoriasis seem to abound but the issue of durability of results is relevant to patients and prescribers. This year, long-term data on secukinumab durability are available to help guide clinical decision-making. Finally, the risk of non-melanoma skin cancer (NMSC) and other cutaneous malignancies has been further studied in order to better understand what contributory role biologics might play in their development.
Certolizumab pegol in pregnancy. Pregnant women represent an important special population in which inflammatory diseases can be particularly difficult to treat due to the risk of transference to the child in utero. CRIB was a prospective, multicenter, post-market, pharmacokinetic study that evaluated the placental transfer of certolizumab pegol (CZP).1 Sixteen pregnant women were entered into and completed the study. The patients had to be at least 30 weeks pregnant, with a diagnosis of an approved CZP indication—psoriatic arthritis, rheumatoid arthritis, Crohn’s disease, or ankylosing spondylitis—and receive the last dose of CZP 35 days or less before delivery. Blood samples for measurement of CZP plasma concentration were taken from mothers, umbilical cords, and neonates at delivery; blood was drawn again from the infants at four and eight weeks post-delivery. At delivery, maternal CZP plasma levels from the 16 patients fell within the anticipated therapeutic range (24.4µg/mL, range 5.0– 49.4). Sixteen neonates were included in the study, but two were excluded from per-protocol datasets, one because of missing data at birth, the other because of implausible pharmacokinetic data. Of the 14 neonates in the per-protocol study, there was no quantifiable level of plasma CZP in 13 infants (<0.032µg/mL) and a minimal level in one (0.042µg/mL). At Weeks 4 and 8, none of the infants had quantifiable plasma levels of CZP. Umbilical cord samples were taken from the 16 patients but one was excluded due to missing data; of the 15 included umbilical cords, three had quantifiable levels of CZP, the highest of which was 0.048µg/mL. Thus, the CRIB clinical trial demonstrated that there was little to no CZP transfer from mother to child and that there was no fetal exposure to CZP during the third trimester of pregnancy. These findings suggest that CZP can be safely administered to pregnant women, when appropriate, without the risk of placental transfer.
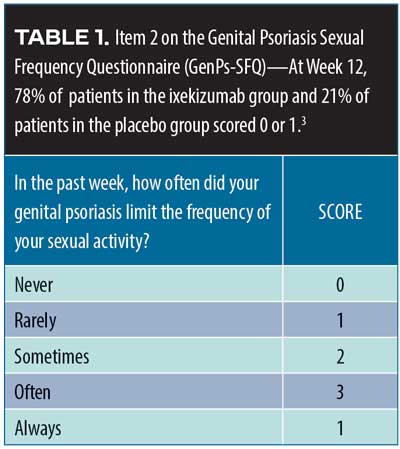
Ixekizumab in genital psoriasis. Genital psoriasis affects up to 63 percent of all patients with psoriaris2 and can be difficult to treat. A recent randomized, double-blind, placebo-controlled Phase IIIb study (IXORA-Q)3 evaluated the potential of ixekizumab for the treatment of genital psoriasis. A total of 149 patients with moderate-to-severe genital psoriasis with body surface area (BSA) greater than one percent were randomized to receive either placebo (n=74) or ixekizumab at recommended doses (n=75). Moderate-to-severe genital psoriasis was defined by the Physician’s Global Assessment of Genitalia (sPGA-G) as having a score of three or higher. The primary endpoints were patients who score zero or one on the sPGA-G, the overall sPGA score, a three-point or more improvement over baseline on the itching numeric rating scale, and response to Item 2 on the Genital Psoriasis Sexual Frequency Questionnaire (GenPs-SFQ), which asked if genital psoriasis limited sexual activity (Table 1). The ixekizumab group received the United States Food and Drug Administration (FDA)-approved psoriasis dosing of 160mg of ixekizumab at Week 0, then 80mg every two weeks through Week 12; the placebo group received placebo on the same schedule. After 12 weeks, patients entered an open-label treatment phase for a total of 52 weeks. Most of the IXORA-Q patients were male (75.8%). The mean age of patients was 43.7± 2.7 years and mean weight was 93.5±24.7kg. Among the participants, 52.3 percent had previously used non-biologic systemic therapies, and19.5 percent had previously used biologic systemic therapies to treat their psoriasis. By Week 12, significantly more patients in the ixekizumab group had clear or almost clear genital-area skin compared to placebo (73.3% vs. 8.1%, p<0.001). Fifty-six percent of ixekizumab patients had completely clear genital-area skin at 12 weeks compared to placebo (5.4%), p<0.001. Patients also reported at Week 12 a clinically meaningful relief from itchiness over baseline with ixekizumab versus placebo (59.7% vs. 8.3%, p<0.001), with a statistically significant difference emerging at Week 2. Item 2 on the GenPs-SFQ (Table 1) found that significantly more patients in the ixekizumab group reported that genital psoriasis never or rarely limited sexual activity at Week 12 versus placebo (78.4% vs. 21,4%, p<0.001) with the difference achieving statistical significance by Week 1 (21.6% vs. 4.8%, p=0.036). Treatment-emergent adverse events (TEAEs) were more frequent with ixekizumab than placebo; most were mild to moderate. The most commonly reported TEAEs with ixekizumab were respiratory tract infection, injection site reactions, oropharyngeal pain, pruritus, back pain, eczema, and hypertension. Severe TEAEs occurred more frequently among the placebo group than the ixekizumab group (4.1% placebo vs. 1.3% ixekizumab). Six patients discontinued the study (6.8% of placebo group and 1.3% of ixekizumab group) because of adverse effects.3
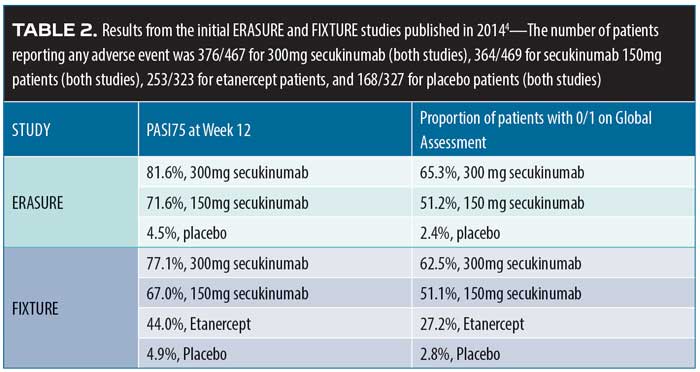
Secukinumab durability. In 2014, the results of two Phase III studies, Efficacy of Response and Safety of Two Fixed Secukinumab Regimens in Psoriasis (ERASURE, N=738) and Full-year Investigative Examination of Secukinumab vs. Etanercept Using Two Dosing Regimens to Determine Efficacy in Psoriasis (FIXTURE, N=1,306) were published.4 Patients with moderate-to-severe plaque psoriasis were recruited. Patients in the FIXTURE study were randomized to receive subcutaneous secukinumab 300mg or 100mg once a week for five weeks, then every four weeks thereafter; etanercept 50mg twice a week for 12 weeks, then weekly thereafter; or placebo. The study objective was to show the superiority of secukinumab over placebo at 12 weeks (defined as Psoriasis Area and Severity Index Score [PASI] of at least 75 percent over baseline [PASI75] and a score of “clear” or “almost clear,” that is, 0 or 1, on a five-point PGA). More patients in the secukinumab group at both doses achieved PASI75 at 12 weeks compared to patients in the placebo group or etanercept group ( Table 2).
The ERASURE and FIXTURE studies were then extended to evaluate long-term control of moderate-to-severe psoriasis following the withdrawal of secukinumab.4 PASI75 responders in the secukinumab 300mg and 150mg groups entered the study and were randomized to be treated for two years with secukinumab (same dose as original study) or placebo. Patients were monitored monthly and could not use any other psoriasis medications. Relapse in placebo patients, defined as greater than 50-percent loss of the previous gain in PASI score reduction, was treated by switching the patients to the secukinumab group. Among the placebo patients, 14 percent and 21 percent of the patients in the previous secukinumab 150mg and 300mg groups, respectively, did not relapse in the first year without medication; in the second year, six percent and 10 percent of the placebo patients who had previously been treated with secukinumab 150mg and 300mg, respectively, did not relapse. The meaning of these data is controversial and should not be interpreted to mean that the typical patient treated with secukinumab will achieve “remitted” control of his or her skin disease. In fact, without an active control group, it is impossible to know if this type of response is typical of many different therapies for psoriasis.
Risk of non-melanoma skin cancer (NMSC) with biologics versus methotrexate. The Psoriasis Longitudinal Assessment and Registry (PSOLAR) database was evaluated to determine the potential risk of NMSC in patients who used biologic therapy compared to those who were eligible for biologic treatment but took methotrexate instead.5 Collection of data was cut stopped on August 23, 2015, and was pooled from 12,090 patients representing 48,870 patient-years of follow-up. Within this group, 6,350 patients used biologics and 432 used methotrexate. Most of the patients in this registry study were Caucasian. This was a well-performed analysis with a comparator group. There were 72 new diagnoses of NMSC (48 basal cell carcinoma [BCC], 24 squamous cell carcinoma [SCC]). This signals a declining rate of BCC with ustekinumab and a trend toward increasing rates of SCC with TNF-inhibitors. That said, the risk of skin cancer with all biologic drugs remains very low, and targeted monitoring of high-risk patients remains the sensible approach.
Risk of malignancy with systemic psoriasis treatment. Using the PSOLAR data, a retrospective, nested case-control analysis6 was conducted on the risk of malignancy for individuals who received systemic psoriasis treatment; this analysis was based on 12,090 registry participants who were followed for 4.2 years, with a maximum follow-up period of 8.2 years. The analysis included patients with newly diagnosed malignancies (other than NMSC) and excluded those with a history of cancer (other than NMSC). Four randomly chosen control patients were matched with each case by age at index date (±6 months), sex, geographical region, and date of enrollment. For the purposes of this study, “exposure” was defined as having received a dose of the study therapy within one year of the index date and stratified by the duration of therapy into three discreet categories: greater than 0 and less than three months; three months to less than 12 months; and 12 months or longer. A total of 252 malignancy cases were found and matched to a total of 1,008 controls. Treatment of psoriasis using methotrexate or ustekinumab for any of the three discreet time periods was not associated with an increased risk of malignancy compared with no exposure to these drugs. However, TNF inhibitors, when used longer term (12 months or longer) were associated with an increased risk of cancer (odds ratio [OR]1.54, 95% confidence interval [CI], range 1.10– 2.15, p=0.01). This risk for TNF inhibitors disappeared when the analysis solely focused on patients receiving these agents as monotherapy. Also, the use of TNF inhibitors for shorter exposure periods (<12 months) was not associated with an increased risk of malignancy.6
References
1. Mariette X, Förger F, Abraham B, et al. Lack of placental transfer of certolizumab pegol during pregnancy: results from CRIB, a prospective, postmarketing, pharmacokinetic study. Ann Rheum Dis. 2018;77(2): 228–233.
2. 1Ryan C, Sadlier M, De Vol E, et al. Genital psoriasis is associated with significant impairment in quality of life and sexual functioning. J American Acad Dermatol. 2015;72(6):978–983.
3. Ryan C, Menter A, Guenther L, et al. Efficacy and safety of ixekizumab in a randomized, double-blinded, placebo-controlled Phase 3b study of patients with moderate-to-severe genital psoriasis. Br J Dermatol. 2018 May 10. doi: 10.1111/bjd.16736. [Epub ahead of print].
4. Langley RG, Elewski BE, Lebwohl M, et al. Secukinumab in plaque psoriasis—results of two phase 3 trials. N Engl J Med. 2014;371(4):326–338.
5. Fiorentino D, Ho V, Lebwohl M, et al. Risk of malignancy associated with biologic therapy and methotrexate (MTX) in the Psoriasis Longitudinal Assessment and Registry (PSOLAR). Poster Abstract B221. Presented at the American Academy of Dermatology. Orlando, Florida. 3–7 March 2017. J Am Acad Dermatol. 2017;76:(6 Suppl).
6. Fiorentino D, Ho V, Lebwohl MG, et al. Risk of malignancy with systemic psoriasis treatment in the Psoriasis Longitudinal Assessment Registry. J Am Acad Dermatol. 2017;77(5):845–854.e845.